Beckwith-Wiedemann syndrome is multisystemic
pattern of congenital anomalies with overgrowth. The most characteristic
clinical features are macroglossia, large birth weight, omphalocele,
visceromegaly, and hypoglycemia. Here we present a case of Beckwith
–Wiedemann syndrome with its major manifestations but without
hypoglycemia. To our knowledge this is the first case to be reported
at King Hussein Medical Center.
JRMS June 2005; 12(1): 38-40Case Report A male newborn, product of normal vaginal delivery following uneventful 38 weeks normal pregnancy to a 25 year primigravida Jordanian mother who is not known to have any medical illness. He had an apgar score of 8/10 at one minute and 10/10 at five minutes.
Physical examination revealed an active pink baby with the following anthropometric measurements: Body weight 3.8 kg (>90th centile); length 52 cm (>90th centile); head circumference 34 cm (50th centile). The baby had impressively large tongue protruding out of the oral cavity and about 5X5 cm omphalocele with loops of intestine inside it, (Fig. 1 and 2).
These findings raised the possibility of Beckwith-Wiedemann syndrome (BWS). Detailed examination showed unusual linear creases and over folded helices of both ears (Fig. 3) but no other dysmorphic features. He had an ejection systolic murmur grade II/VI over the left lower sternal border; 2D echocardiograph showed a tiny perimembraneous ventricular septal defect (VSD).
Genitourinary evaluation showed undescended right testicle but otherwise normal male genitalia. The baby had no other abnormality and his general condition was stable; he was started on early bottle feeding and his blood glucose level was monitored hourly for the first eight hours, then every four hours for forty eight hours without evidence of hypoglycemia or hyperinsulinism. Thyroid function test was normal.
Surgical consultation was requested for the omphalocele, which was operated uneventfully on the third day of life. Abdominal ultrasound showed normal viscera with mild liver enlargement.
The baby is now six months old; his follow up so far has been regular, with normal development, although his weight and length still go parallel to the 90th centile and still having large tongue but without causing feeding or respiratory difficulties (Fig. 4).
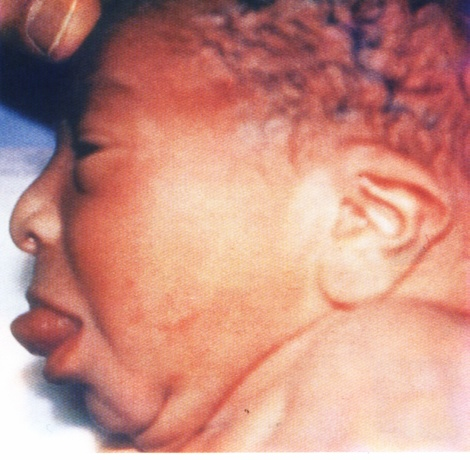 Fig. 1:
Fig. 1: Macroglossia in Beckwith-Wiedemann syndrome.
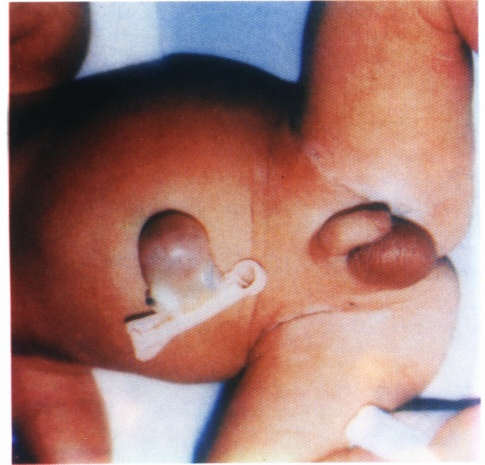 Fig. 2:
Fig. 2: Omphalocele in Beckwith-Wiedemann syndrome.
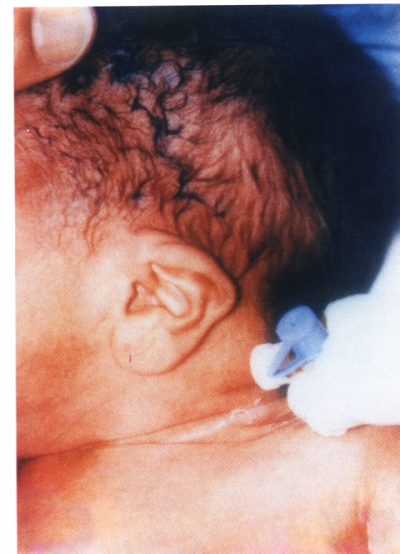
Fig. 3: Linear creases.
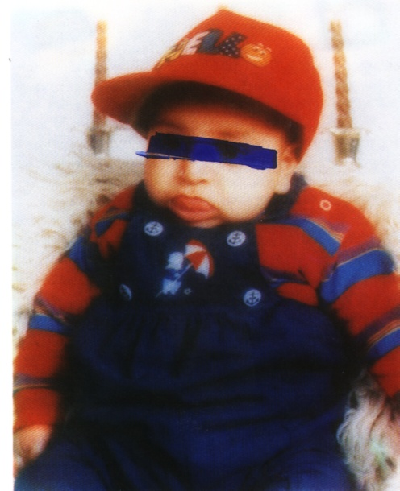

Fig. 4:After 6 months
DiscussionBeckwith-Wiedemann syndrome is a congenital overgrowth syndrome first described by Beckwith in 1963. The incidence of BWS is about 1:13700 births, with an equal sex distribution (1). It is a clinically and genetically heterogenous disorder. The existence of mild forms of BWS probably underestimates this incidence. The phenotype of BWS is likely to result from an imbalance of a number of critical genes at chromosome 11p 15.5. At this location, genetic imprinting with the loss of maternally expressed tumour and / or growth supressor genes - for example, p57 and H19 – or duplications and uniparental disomy of paternally expressed growth promoter genes - for example, insulin-like growth factor II – have been implicated in BWS. In BWS, 85% of cases are sporadic and 15% are autosomal dominant (2).
Elliott and Maher identified criteria that are present in our case for post natal diagnosis of BWS, the three major features are pre- and / or post-natal overgrowth (>90th centile), macroglossia and abdominal wall defects, with minor defects of characteristic ear signs (ear lobe creases or posterior helical pits), facial nevus flammeus, hypoglycemia, organomegaly and hemi-hypertrophy. Postnatal diagnosis is based on either 1- three major features or 2- two major plus three or more minor features (3).
Reish et al made it possible by ultrasound and cytogenetics analysis of the fetus and both parents to diagnose the condition parentally. Constant ultrasound findings include fetal overgrowth, polyhydramnios, enlarged placenta, and specifically a distended abdomen. These signs usually develop after twenty-two weeks of gestation (4).
As in our case, the dysmorphic features become less apparent during childhood, although the macroglossia may cause feeding, speech problems, and /or obstructive apnoea that may develope in this patient later, so surgical tongue reduction may be required in severe cases (5).
Overgrowth is most marked in the first few years and is associated with an advanced bone age. It tends to slow down in late childhood, and most adults are <97th centile. BWS must be followed for the neoplasia that may occur in 5% of patients, most commonly is Wilms’ tumour, especially, those children with hemihypertrophy (25%). By adolescence, the majority led a normal life. There is controversy about abdominal tumour screening which some centers advocate should be performed by regular abdominal palpation and others by regular ultrasound examination or both (6).
Hypoglycemia was not reported in our case, while the incidence of hypoglycemia is about 50%. In the majority (80%) it is mild and asymptomatic, requiring extra feeds or intravenous dextrose only and in 20%, the hypoglycemia is prolonged more than a week and difficult to control.
Hypoglycemia tends to ameliorate spontaneously after several months, although it has been documented into the third year of life .Our patient has normal development because he did not develop hypoglycemia, and intellectual impairment is mostly associated with hypoglycemia. The common feature of all the case reports of hypoglycemia in BWS is hyperinsulinism (which is normal in our patient), with inappropriate insulin secretion in the presence of hypoglycemia. The histological evidence is consistent with diffuse islet and B cell hyperplasia and hypertrophy. A reduction in somatostatin-producing cells has been noted (7).
The treatment of hypoglycemia in BWS should be prompt, with vigilant control of blood sugar levels. Thus, adequate carbohydrates should be provided (oral and intravenous), followed by oral agents such as diazoxide, chlorothiazide, and nifedipine. The use of parenteral agents such as glucagon and octreotide may be necessary if oral treatment fails. Finally pancreatic surgery may be required if maximum medical treatment fails to control hypoglycemia (8,9).
References1.
Munns CFJ, Batch JA. Hyperinsulism and Beckwith-Wiedemann syndrome. Arch Dis Child (Fetal Neonatal Ed) 2001; 84: 67-69.
2.
Jones KL. Beckwith-Wiedemann Syndrome. In: Jones KL, editor. Smith’s recognizable pattern of human malformations 5th ed. W.B. Saunders. Philadelphia 1988; 136-1137.
3.
Elliott M, Maher ER. Beckwith-Wiedemann syndrome. J Med Genet 1994; 31: 560- 564.
4.
Reish O, Lerer I, Amiel A, et al. Wiedemann-Beckwith syndrome: Further prenatal characterization of the condition Am J Med Genet 2002; 107: 209- 213.
5.
Kotoku R, Kinouchi K, Fukumitsu K, et al. A neonate with Beckwith-Wiedemann syndrome who developed upper airway obstruction after glossopexy. Masui 2002; 51: 46-48 (Abstract)
6.
Sperling MA, Menon RK. Hyperinsulinemia hypoglycemia of infancy. Endocrinol Metab Clin North Am 1999; 28(4): 695-707.
7.
Lteif AN, Schwenk WF. Hypoglycemia in infants and children. Endocrinol Metab Clin North Am 1999; 28(4): 619- 649.
8.
Stanley CA. Hyperinsulinism in infants and children. Pediatr Clin North Am 1997; 44: 363-374.
9.
De Lonlay-Debeney P, Poggi-Travert F, Fournet JC, et al. Clinical features of 52 neonates with hyperinsulinism. NEJM 1999; 340: 1169-117