Objective: To describe a single center experience of a rare disease, with special emphasis on clinical presentation, treatment options and prognosis.
Methods: We retrospectively reviewed the files of 12 patients discharged with the diagnosis of chronic granulomatous disease between July 1997 and June 2004 in the pediatric immunology clinic at King Hussein Medical Center.
Results: Twelve patients, seven males, and five females were identified. They aged between 13-84 months with a mean age of 29 months at the time of diagnosis. The median duration of follow-up was 33 months. Eight patients were alive and four had died. Family history of Chronic granulomatous disease was positive in 10 patients. The median duration of delay in diagnosis was 27 months. Two patients had Aspergillus chest infection; one tuberculosis meningitis and two had multiple liver abscesses. A microorganism isolates included Aspergillus species, Salmonella, Staphylococcal aureus, and Pseudomonas aeruginosa. None of our patients had non-infectious complications. All patients were failing to thrive at the time of diagnosis. Most of them achieved acceptable growth after 24 months of treatment. All patients received Trimethoprium-sulphamethoxazole and Itraconazole. None of our patients underwent bone marrow transplant. The duration of hospital admission was significantly decreased after treatment from a median of 19 weeks before to two weeks after treatment.
Conclusion: Awareness of general pediatricians toward early diagnosis was suboptimal. Every effort should be made to improve the laboratory diagnosis by using more specific tests. Anti-microbial prophylaxis improved the prognosis of chronic granulomatous disease significantly. Interferon-γ and bone marrow transplant should be considered for the patients with poor response to daily antimicrobial prophylaxis.
Key words: Chronic, Granulomatous, Disease, Diagnosis, Complications, Treatment.
JRMS Dec 2006; 13(2):24-29
IntroductionChronic granulomatous disease (CGD) is uncommon congenital Immunodeficiency with the incidence of one in 250, 000 individual (1,2). It is caused by a profound defect in an oxygen metabolic burst that normally accompanies phagocytosis in all myeloid cells (1,2).
As a result of the failure to activate the respiratory burst in their phagocytes, the majority of CGD patients suffer recurrent infections, the most common of which are pneumonia, lymphadenitis, cutaneous and hepatic abscess, osteomyelitis, and septicemia (1,2).
These severe infections usually become apparent during the first year of life and are caused predominantly by Staphylococcus aureus, Aspergillus species, enteric gram-negative bacteria, Serratia marcescence, and Pseudomonas cepacia (1-3). In addition, CGD patients have diffuse granulomas that can become sufficiently large to cause obstructive or painful symptoms in esophagus, stomach, biliary system ureters, or urinary bladder (4,5). CGD is inherited as X-linked recessive in 65 % of cases and autosomal recessive in 35 % (2-3).
Conventional treatment consists of lifelong antiinfective prophylaxis with antibiotics such as cotrimoxazole and antimycotics such as itraconazole and /or with interferon gamma (6,7,8). Despite these measurements the annual mortality is still between 2 (autosomal recessive CGD) to 5 (X-linked CGD) percent (8). An alternative to conventional treatment, hematopoietic stem cell transplantation (HSCT), should be done early before patients develop any irreversible organ damage (9-15). We reviewed 12 files of patients discharged with the diagnosis of CGD.
MethodsAll files reviewed were of patients discharged with the diagnosis of chronic granulomatous disease (CGD) between July 1997 and June 2004 at the pediatric immunology clinic at King Hussein Medical Center (KHMC). The files were reviewed for demographics, age at presentation, clinical course after diagnosis, growth achievement, antimicrobial prophylaxis, and laboratory parameters. We initially diagnosed all cases by means of the phorbol myrisate acetate-stimulated nitroblue tetrazolium test (NBT), and only one of them was confirmed by flowcytometry burst test using Dihydrorhodamine (DHR) dye (Fig. 1). Mutation analysis was not available in Jordan.
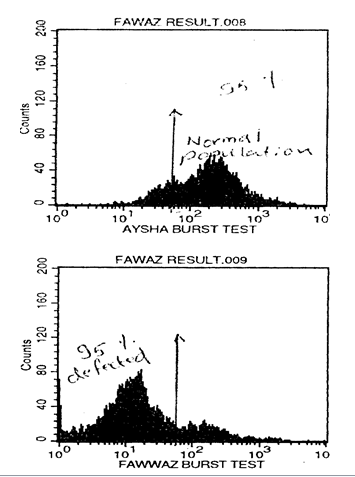 Fig. 1:
Fig. 1: A. Flow Cytometry-based burst test display dihydrodamine (DHR) fluorescence in a patient with CGD. A: positive control (normal), B: Patient result shows abscence of fluorescence activity. Adapted with permission from Al-Wahadneh et al. JBMS 2001; 13(2): 101-105.
Results
Twelve CGD patients (7 males and 5 females) aged between 13-84 months (median 31 months) were identified between 1997 and 2004. The median duration of follow-up was 33 months. They all are permanent residents of Jordan and have Arabic racial pattern. These patients belonged to 6 families, and the parents of all patients were consanguineous. Most of our patients were diagnosed as late as 27 months after initial presentation.
None of them was diagnosed before the onset of symptoms or in the neonatal period even in those who had positive family history of CGD. Tables I and II show clinical and laboratory features at initial presentation. Four patients died (33.3%) during the follow up because of recurrent infections and complications, including acute disseminated aspergillosis (in a 4-year-old boy); he showed typical chest CT scan findings of acute pulmonary Aspergillosis (Fig. 2), sepsis after the rupture of multiple bacterial liver abscess (in a 9-year-old girl), sepsis after pneumonia (in a 9-month-old boy), and acute miliary tuberculosis (in a 5-year-old boy).
One patient had subacute bacterial meningitis. Cultures for bacterial, mycobacterium and asperigillus infections were negative. He was not vaccinated by BCG. The diagnosis of tuberculous meningitis was made because of Cerebrospinal fluid (CSF) lymphocytosis, poor response to antibacterial therapy, dramatic response to 9 months of triple anti-tuberculosis regimen and proved contact with patient with active tuberculosis.
He recovered with moderate communicating hydrocephalus (Fig. 3). Another female patient presented at 5 months of age with severe respiratory distress. Chest X-ray and CT scan showed mass-like lesion with rib involvement (Fig. 4). The mass was surgically removed, and proved to be aspergilloma containing Aspergillus fumigatus. She continued to be well on trimethoprium- sulphamethoxazole and Itraconazole prophylaxis.
A newly diagnosed 9- year- old boy presented with multiple liver abscesses of variable sizes (Fig. 5). Surgical drainage was performed due to failure of response to medical treatment. None of our patients had non-infectious complications. All patients were failing to thrive at the time of diagnosis. Five patients (42%) achieved acceptable weight and height gain after 2 years. All patients received trimethoprium-sulphamethoxazole (10mg/kg/day) and Itraconazole (5mg/ kg/day) prophylaxis for bacterial and Asperigillus at the time of diagnosis.
They all tolerated treatment well. The median duration of hospital admission for major infection was two weeks (ranges 2-5 weeks) with prophylactic treatment compared to 19 weeks (range between 12-29 weeks per patient per year) before treatment.
None of our patients underwent bone marrow transplant and none of them received Interferon-gamma as prophylaxis or as treatment agent. Purified protein derivtive (PPD) was negative in all patients at initial presentation. The microorganisms isolated were Aspergillus species, Salmonella species, Staphylococcal aureus and Pseudomonas species. All patients had high ESR and CRP during exacerbation, which are decreased significantly in most patients after treatment. All patients remained anemic despite tonic supplement.
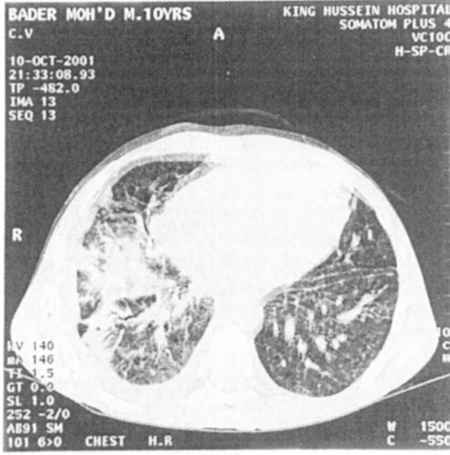 Fig. 2:
Fig. 2: Chest CT scan of a male with CGD and Aspergillus infection
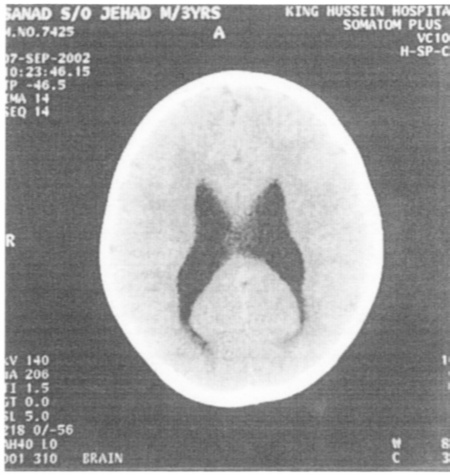 Fig. 3:
Fig. 3:Brain CT scan shows moderate dilatation of the ventricular system associated with persistent extravasation of the CSF. Appearances are those with moderate communicating hydrocephalus.
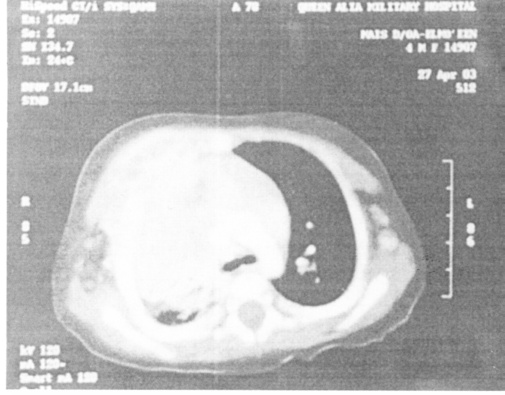
Fig. 4. Chest CT scan of a female with CGD and Asperigillus infection (Asperigelloma) spread contagiously to ribs.
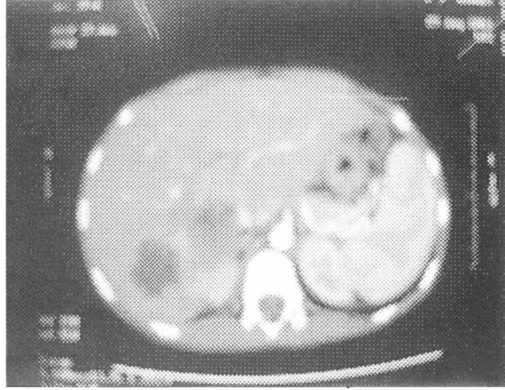 Fig. 5.
Fig. 5. Abdominal CT scan showing Hepatic abscesses.
Discussion
To the best of our knowledge, this report was the first on Jordanian CGD patients comprising the clinical data of 12 patients in a single center during 8-year period. Male patients constituted 58% of our patients.
The male to female ratio in our study (1.4:1) was lower than that in other studies, which may be due to higher autosomal inheritance related to increased consanguinity of families in Jordan. Two out of six families showed X-linked mode of inheritance, while the other four showed autosomal recessive pattern.
These findings were in keeping with similar studies in Iran and Tunisia, 2.4:1 and 1.8:1 respectively (16,17). However, the relative preponderance of X-Linked cases compared with autosomal recessive CGD cannot be precisely defined in our series. We do not have mutation analysis in our laboratory, and although NBT test also proved to be a powerful tool in defining the genetic of CGD, we do not do NBT routinely for mothers of affected males (3,4). Even though, family history was positive in 10 (83%) of our patients, none was diagnosed as CGD before the onset of symptoms compared with Goldblatt who referred to 30 % of his cases were diagnosed before onset of symptoms depending on positive family history (4).
Although half of patients with CGD were diagnosed before the age of 5 years, but as with many rare diseases, there is always delay between the onset and time of diagnosis and 20% of cases were not diagnosed until the 2nd decade of their life (4,16,17).
We were late by 27 months compared with others who had a median delay of 13 months in developed countries (3,4,16,17). The average diagnostic delay in one of the developing countries (Iran) was 45 months (16). These data showed the lack of awareness of CGD among the general practitioners and pediatricians in developing countries (4,16,17). Lymphadenopathy was the most common presentation (83%), which is similar to the percentages in previous studies (4,16,17).
In some other studies, respiratory manifestations were the most common (3,4,16,17). It was the initial presentation in 60% of our patients. Pulmonary tuberculosis was documented in one patient who died of acute miliary Tuberculosis without previous BCG vaccination (16-19).
The incidence of pulmonary tuberculosis in patients with CGD is variable; some reported as high as 32% of cases, while others found that this infection did not play a significant role among the CGD patients (4,16-19).
This might be due to the difference in exposure to Mycobacterium tuberculosis in different countries (4,16-19). Asperigillus pneumonia had been identified in two patients (16%); this is in keeping with other reports that found that the incidence of Asperigillus pneumonia ranged between 7-10% (4,16,17). It might be attributed to the improvement in controlling bacterial infections by antibiotic prophylaxis, which makes the invasive fungal infections; especially the ones of Asperigillus species are now the most important causes of morbidity and mortality in CGD in spite of Itraconazole prophylaxis (4,16,17). Cellulitis and osteomyelitis were not documented in our study, in contrast with other researchers who reported a prevalence of 20% in their patients’ cohort (4,16,17).
The differences in the relative prevalence of infections between different series may be related to a variety of factors, including the number of patients reported, the
changing clinical expression of the disorder over time, and the way in which information on specific infections was reported (4,16,17). Despite data to show that good preventive treatment improves survival and quality of life in children, CGD is still associated with high morbidity and mortality (4,8).
There were 4 deaths in our cohort; all of them were found non-compliant to anti-microbial treatment. Goldblatt reported that the incidence of severe infections with prophylaxis was significantly low, at 0-11 per patient year follow up (4).
We also achieved a significant decrease in incidence of severe infection; it decreased by 78%. We believe that the improvement in Goldblatt and our series is due to proper coverage of both bacterial and fungal microorganism [4,16-20).
The use of interferon gamma was advised only when patients develop severe deep infection despite oral treatment (4-21). None of our patients had non-infectious complication compared with other cohorts (3,4).
This is in keeping with Finn et al who did not report any non-infectious complications and Goldblatt and Cohen; who reported less than 10% of their multi-center cohorts (3,4).
It could reflect either a historical failure to distinguish infections from non-infectious symptoms or a rare increase in the non-infectious complication, in addition to the fact that our patients were followed for a relatively short period of time, where most inflammatory complications occurred in the second decay of life (4,16,17). Prophylactic antimicrobials are the main stay of treatment for patients with CGD (3-7,21).
However, serious breakthrough infections are common (3-7,16,17). These are encouraging trials of bone marrow transplant early in life, before the appearance of severe infections or irreversible tissue damage, as a curative treatment with some limitations (9,10,14,15).
We concluded that awareness of general pediatricians toward early diagnosis was suboptimal. Every effort should be made to improve the laboratory diagnosis by using more specific tests. It seems necessary for future studies to the genotype of our CGD patient and improve the carrier screening of X-linked cases in certain families for early diagnosis and prevention of severe complications.
Bacterial and fungal prophylaxis improved the prognosis of CGD significantly. Experience with Interferon-γ and bone marrow transplant was substantial and should be encouraged for patients with poor response regardless of daily prophylaxis.
Table I: Demographic, clinical features, and treatment options of 12 patients
|
Index/ Patient number
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Age (months)
|
13
|
24
|
20
|
24
|
84
|
24
|
36
|
20
|
9
|
15
|
98
|
96
|
|
Sex
|
M
|
M
|
F
|
M
|
M
|
M
|
F
|
F
|
F
|
M
|
F
|
M
|
|
Age of onset (months)
|
3
|
9
|
6
|
9
|
10
|
9
|
6
|
6
|
1
|
3
|
64
|
1
|
|
Disease duration before diagnosis (months)
|
10
|
15
|
14
|
15
|
74
|
15
|
30
|
14
|
5
|
12
|
34
|
95
|
|
Family history
|
-
|
+
|
-
|
-
|
+
|
-
|
-
|
-
|
+
|
-
|
+
|
+
|
|
Lymphadenitis
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
|
Diarrhea
|
+
|
+
|
-
|
+
|
+
|
+
|
+
|
-
|
+
|
-
|
-
|
-
|
|
Perianal abscess/fissure
|
+
|
+
|
-
|
-
|
+
|
+
|
+
|
-
|
+
|
-
|
+
|
+
|
|
Liver abscess
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
+
|
-
|
+
|
+
|
|
Asperigillosis
|
+
|
-
|
-
|
-
|
-
|
-
|
-
|
?
|
-
|
-
|
-
|
?
|
|
Recurrent respiratory infection
|
+
|
+
|
+
|
+
|
+
|
-
|
-
|
+
|
+
|
+
|
+
|
-
|
|
Granulomatous colitis
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Sepsis(Salmonella)
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
+
|
-
|
-
|
-
|
+
|
|
Staphylococcal skin infection
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
-
|
+
|
|
Failure to thrive
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
|
Granulomatous cystitis/urethritis
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Recurrent mouth ulcers
|
+
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Gingivitis
|
+
|
+
|
+
|
-
|
-
|
+
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Pericarditis
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Upper gastrointestinal obstruction
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Antibiotic prophylaxis
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
-
|
+
|
|
Anti-fungal prophylaxis
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
+
|
-
|
+
|
-
|
+
|
|
Interferon treatment
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Bone marrow transplant
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
+: Present -: Absent ?: not proved radiologically or microbiologically
Table II: Immunological laboratory tests.
|
Test/Patient number
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
IgG
|
H
|
H
|
H
|
H
|
H
|
H
|
H
|
H
|
H
|
H
|
H
|
H
|
|
IgA
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
IgM
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
NBT(resting/stimulated %)
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
0%
|
|
FCM-burst test
|
Abn
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
|
Isoheammaglutinn
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
CD3
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
N
|
|
CD4
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
N
|
|
CD8
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
N
|
|
CD19
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
N
|
|
CD4:CD8
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
N
|
|
N
|
|
CH50
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
|
HIV
Total WBC/PMN
|
-
I / I
|
-
I / I
|
-
I / I
|
-
I / I
|
-
I /I
|
-
I / I
|
-
I / I
|
-
I / I
|
-
I / I
|
-
I / I
|
-
I/ I
|
-
I / I
|
|
H: high, N: normal, Abn: abnormal, NA: not available, -: negative. I: increased
References
1.
Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defect in phagocytes. N Engl. J Med 2000; 343(23): 1703-1715.
2.
Fischer A. Human Primary Immunodeficiency diseases: A perspective. Nature Immunology 2004; 5(1): 23-30.
3.
Roos D. Curnutte JT Chronic granulomatous disease. In: Ochd HD, Edvard Smith C.I, Puck JM, editors Primary Immunodeficiency disease: A molecular and genetic approach. 1st edition. New York: Oxford University Press 1999; 353-374.
4.
Cale CM, Jones AM, Goldblatt D. Follow up of patients with chronic granulomatous disease diagnosed since 1990. Clinical and experimental immunology 2000; 120: 351-355.
5.
Segal BH, Leto TL, Gallin JI, et al. Genetic, biochemical, and clinical features of chronic Granulomatous Disease. Medicine 2000; 79(3):170-300
6.
Gallen JI, Alling DW, Malech HL, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl. J Med 2003; 348(24): 2416-2422.
7.
Mouy R, Veber F, Blanche S, et al. Long-term Itraconazole prophylaxis against Aspergillus in Thirty-Two patients with chronic granulomatous disease. J Pediatr 1994; 125: 998-1003
8.
Finn A, Hadzic N, Morgan G, et al. Prognosis of chronic granulomatous disease. Arch Dis Child 1990; 65: 942-945
9.
Ozsahim H, Von Planta M, Muller I, et al. Successful treatment of invasive asperigillosis in chronic granulomatous disease by bone marrow transplant, granulocyte colony stimulating factor-mobilized granulates, and liposomal Amphotericin-B. Blood 1998; 92: 2719-2724.
10.
Bielorai B, Toren A, Wolach B, et al. Successful treatment of invasive asperigillosis in chronic granulomatous disease by granulocyte transfusion followed by peripheral blood stem cell transplantation. Bone Marrow Transplant 2000; 26: 1025-1028.
11.
Negler A, Ackerstein A, Kapelushnik J, et al. Donor lymphocyte infusion post-nonmyeloablative allogeneic peripheral blood stem sell transplantation for chronic granulomatous disease. Bone marrow Transplant 1999; 24: 3339-342.
12.
Salvin S, Nagler A, Naparstek, E, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for treatment of malignant and nonmalignant hematological diseases. Blood 1998; 91: 756-763.
13.
Horwitz ME, Barrett J, Brown MR, et al. Treatment of chronic granulomatous disease with Nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med 2001; 344: 881-888.
14.
Kamani N, August CS, Cambell DE, et al. Marrow transplantation in chronic granulomatous disease: An update with 6-year follow-up. J Pediatr 1998; 113: 697-700.
15.
Leung TF, Chik KW, Li CK, et al. Bone marrow transplantation for chronic granulomatous disease: Long-term follow-up and review of literature. Bone marrow Transplant 1999; 24: 567-570.
16.
Movahedi M, Aghamohammadi A, Rezaei N, et al. Chronic Granulomatous Disease: A clinical survey of 41 patients from the Iranian primary Immunodeficiency registry. Int Arch allergy Immunol 2004; 134: 253-259.
17.
Mateos MA, Alvaro M, Giner M, et al. Chronic granulomatous disease: Six new cases. Allergologia et immunopathlogia 1998; 26(5): 241-249.
18.
Moss W, Ledeman H. Immunization of the immunocompromised host. Clinical focus on primary immune deficiencies 1998; 1(1): 1-8.
19.
Urban C, Beker H, Mutz I, Fitsch G. BCG-infection in chronic granulomatous disease (author tranasal). Klin Padiatr 1980; 1: 13-18. (Abstract)
20.
Fleishmann J, Church JA, Leher RI. Primary Candida meningitis and chronic granulomatous disease. Am J Med Sci 1986; 291(5): 334-341. (Abstract)
21.
Augus S, Spektor S, Israel Z. CNS granulomatosis in a child with chronic granulomatous disease. Br J Neurosurg 2000; 14(1): 59-61. (Abstract)
22.
Mouy R, Seger R, Bourquin JP, et al. Interferon gamma for chronic granulomatous disease. N Eng. J Ed 1991; 325: 1516.
23.
Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatuos disease with myeloablative conditioning and unmodified hemopoietic allograft: a survey of the European experience (1985-2000). Blood 2002; 100: 4344-4350.