Abstract
Objectives: To describe the demographic
characteristics and clinical presentation of 37 patients with Wilson's disease
followed up at the Pediatric Gastroenterology Clinic.
Methods: A specially designed data collection form was used to collect the
relevant data; Medical history and a thorough clinical examination for patients
who were diagnosed with Wilson's disease during the period between February 2000
and October 2010 at King Hussein Medical
Center, Amman, Jordan
was done. Laboratory investigations include ceruloplasmin level, liver enzymes,
albumin, prothrombin time, partial thromboplastin time, international
normalized ratio, complete blood count, urine analysis, abdominal
ultrasound and liver biopsy. Simple descriptive statistics (frequency and
percentage) were used to describe the study variables.
Results: A total of 37 patients
diagnosed as Wilson's disease with age ranges between two and 13.5 years were included
in this descriptive review. Out of 37 patients, 19 (51%) were males and 18 (49%)
were females. Patients with affected siblings were 29 (78%). Central nervous
system involvement was found among 9 (24.3%) patients. The commonest presenting
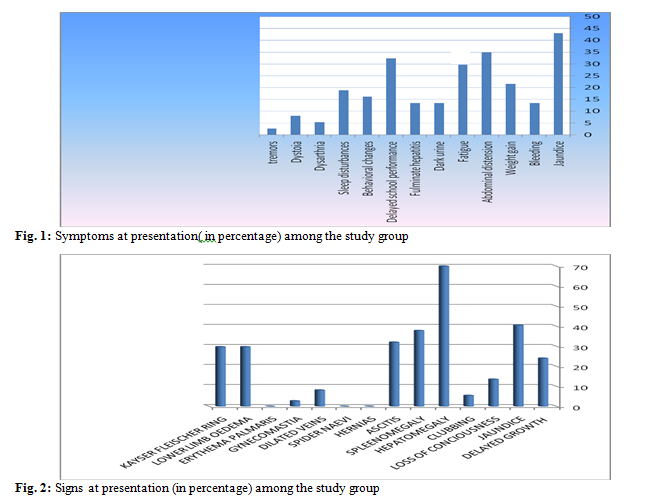
symptoms were jaundice (n=16, 43%), abdominal distension (n=13, 35%), fatigue
and delayed school performance (n=12, 32.4%). The most common clinical findings
were hepatomegaly (n=26, 70%), jaundice (n=16, 43%), splenomegaly (n=14, 37.8%),
Kayser-Fleischer ring (n=11, 29.7%), and lower limb edema (n=11, 29.7%) respectively.
Low ceruloplasmin level was found in 34 (92%) patients, high liver enzymes in
23 (62%) patients, hemolytic anemia in 13 (35%) patients successively. Twenty-four
hour urine collection average copper post D-penicillamine challenging test was
above 230µg/dl. The most common ultrasound findings were hepatomegaly, abnormal
echogenecity, splenomegaly and ascitis. Liver biopsies commonly showed liver
fibrosis, however fatty liver changes, hepatosteatosis and liver cirrhosis were
the least common finding.
Conclusion: Family screening is needed once a child in the family is diagnosed. Full
investigations to rule out Wilson's disease should be performed in any patient
with unexplained elevation of liver enzymes, hepatomegaly, hemolytic anemia,
jaundice or neurological/behavioral disturbances.
Key words: Jaundice, Hepatomegaly, Splenomegaly,
Kayser-Fleischer ring, Wilson's disease
JRMS June 2013; 20(2): 6 -9 / DOI: 10.12816/0000083
Introduction
Wilson's disease is a disorder of copper metabolism characterized by
degenerative changes in the liver, brain, cornea and kidney. It is an autosomal
recessive disease (chromosome 13 q gene: copper binding p-type ATPase, ATP 7B).(1,2)
It is not a common disease
worldwide, its incidence is 1/100,000-1/500,000 live births according to
American studies.(3) Although a Greek and an Indian study
showed a higher incidence of the disease (1 in 30,000) with an average age of
hepatic symptoms being 10-14 years.(1,4) Wilson's disease has a wide spectrum of
clinical manifestations, although some patients may be entirely asymptomatic
with delayed diagnosis.(1) This study was conducted to
determine the demographic characteristics and clinical presentation of 37
patients with Wilson's disease, followed up at the Pediatric Gastroenterology Clinic.
Methods
This is a descriptive retrospective review of the medical records for
all patients who were clinically confirmed and laboratory diagnosed cases with Wilson's
disease at King Hussein Medical
Center during the period
between February 2000 and October 2010, in the Pediatric Gastroenterology
Clinic. We enrolled 37 patients in our study, aged 2-13.5 years, reviewed and
followed up at the Pediatric Gastroenterology Clinic, focusing at symptoms on
first presentation, family history, clinical examination, laboratory data,
radiological findings and liver biopsy. A
full detailed history was taken for each patient and a complete clinical
examination was performed. Laboratory investigations included ceruloplasmin
level, liver function tests, albumin, partial thromboplastin time (PTT), International
Normalized Ratio (INR), prothrombin time (PT), complete blood count
(CBC) with blood film and reticulocytes, and urine analysis. Abdominal Ultrasound
was performed for all our patients and finally, liver biopsy was performed for a
group of patients only and was not done for the others because of significant
coagulopathy or family refusal of liver biopsy. Ferenci's diagnostic score of Wilson's disease
was used (8th international conference of Wilson's disease and
Menkes disease, Leipzig/Germany in April 2001).(5) A
specially designed data collection form was used to collect the relevant data.
Results
The male to female ratio was almost 1:1 (51% and 49% respectively). Family history was positive in 29 (78%) patients,
CNS involvement in 9 (24.3%) patients. The commonest presenting symptoms were jaundice
(n=16, 40%), abdominal distension (n=13, 35%), fatigue and delayed school
performance (n=12, 32.4%) (Fig. 1). The most common clinical findings were
hepatomegaly (n=26, 70%), jaundice (n=16, 43%), splenomegaly (n=14, 37.8%),
Kayser-Fleischer ring (n=11, 29.7%), and lower limb edema (n=11, 29.7%) respectively
(Fig. 2). Low ceruloplasmin level was
found in 34 (92%) patients, high liver enzymes in 23 (62%) patients, hemolytic
anemia 13 (35%) patients successively. Ceruloplasmin level was never low in 100%
of cases in international studies.(1,6,8) Concerning 24 hour urine collection: copper
post D-penicillamine challenging was above 230 micrograms/24hr. Ultrasonography
was performed in all our patients, 64% of which had abnormal findings of which
the commonest finding was hepatomegaly (50%), abnormal echogenicity of the
liver (26%) followed by splenomegaly (20%) and ascitis (13%). Finally, liver
biopsy was performed in 15 (40%) patients and showed liver fibrosis in 54%,
fatty liver in 45%, hepatosteatosis in 18% and liver cirrhosis in 18%.
Discussion
We noticed a high percentage of affected relatives due to consanguinities
especially in Al-Tafeelah and Al-Mafraq district areas. The ceruloplasmin level was low in 92% of
patients compared to 88% in a Brazilian study,(6) and 93% in
Nazer et al. study.(7) In several patients, liver
biopsy may be needed to assess the extent and severity of liver disease. Neurological
features may be the presenting manifestation of Wilson's disease even in the
absence of clinical evidence of hepatic involvement.(4) We compared our finding with Nazer et al.'s
study results which was conducted at King's College Hospital/London. They found
that the commonest symptoms of presentation of Wilson's disease were lethargy
and anorexia (70%), jaundice (56%), and abdominal pain(48%).(8) Patients
below five years of age were diagnosed during family screening(affected
siblings) and they were asymptomatic. In a recent study from Karachi/Pakistan,(9)
viral causes were found to be the most common factor for fulminant hepatic failure in
children (37 patients out 50) which makes 74% of the total, 56% of them had
hepatitis A virus, and 18% had hepatitis B virus. The remaining 26% of the
total (13 patients) were negative for acute serology of hepatotrophic viruses, of
which 8% had Wilson's disease, 2% with autoimmune hepatitis and finally the
etiology could not be established in 16% of cases. In our study, 9 patients
(24.3%) were found to have CNS involvement, which was significantly less than
the international numbers. This could be
partly explained by the presence of different methods to identify the CNS
involvement. In our practice, we do not perform MRI as a routine test. Kayser-Fleischer
ring has been found in 11 patients (29.7%), compared with 41% in a Brazilian
study,(4) 38% in a Greek study(1) at Athens
University and in 32% in an Indian study.(10) Some striking
findings were found in our study concerning microscopic haematuria found in 14
patients (37%) and albuminuria in 6 patients (16%). The Wilson's disease gene
is expressed in kidney tissue,(11) and patients may resemble
those with Fanconi syndrome, urolithiasis, also haematuria and proteinuria can occur
before treatment as part of the disease process and after therapy as adverse reaction to
D-penicillamine therapy.(12) Cases presented with negative
direct Coomb's test, haemolytic anemia in our study constituted 35% (13
patients), compared to 15 % in Rahil Shah study in 2009.(13) The
copper in tissue assay can produce false negative results in children since it
is dependent on sample size, length of time during which the metal has been
accumulating and a fact that it may be irregularly distributed.(4)
In international studies, histopathological findings of hepatic
involvement may vary from steatosis to end stage liver cirrhosis. In Nazer et
al.'s study,(8) the majority of their patients who underwent
liver biopsy, were found to have micronodular cirrhosis, followed by chronic
active hepatitis with moderate piece-meal necrosis.(8) In our
study, the most common histopathologic finding was liver fibrosis followed by
fatty liver (fatty liver can very rarely present in nodular pattern.(14)
In a Greek study,(1) fibrosis was found in a 4 months old
infant, and inflammation was found in a 23 months old child, suggesting that
serious histological changes may develop during the early stages of the disease.
Conclusion
The commonest clinical presentations of Wilson's disease in Jordan were
Jaundice and hepatomegaly. Family screening is important once you diagnose Wilson's
disease. Full investigations to rule out Wilson's disease should be performed
in any patient with unexplained high liver enzymes, hepatomegaly, haemolytic
anemia or neurological/behavioral disturbances. Finally, haemolytic anemia,
microscopic haematuria were strikingly higher in our patients than that found
in international studies.
References
1.Manolaki N; Nikolopoulou G; Daikos G L, et al. Wilson's disease in children: Analysis of 57 Cases. Journal of
Pediatric Gastroenterology & Nutrition Jan 2009; 48 (1): 72-77.
2.Merle U, Weiss K H, Eisenbach C, et al. Truncating
mutations in the Wilson's disease gene ATP7B are associated with very low serum
ceruloplasmin oxidase activity and an early onset of Wilson's disease. BMC
Gastroenterology 2010, 10:8doi: 10.1186/1471-230x-10-8.
3.Rodolph JA, Balistreri WF. Metabolic diseases of the liver. Nelson text book of pediatrics. 17th
edition.2004, (p1321-1322), USA.
4.Taksande A,
Vilhekar K, Parihar PH. Unusual manifestation
in child of Wilson's’s disease. Journal of Chinese Clinical Medicine
2008, 10; 3-10.
5.Steindl P, Ferenci P, Dienes HP, et al. Wilson's disease in patients
presenting with liver disease, a diagnostic challenge. Gastroenterology, 1997, 113:212-18.
6.Stephania A, Alexandre R, Eleonora F, et al. Wilson's’s
disease in children and adolescents: diagnosis and treatment. Rev Paul
Pediatr 2010; 28(2):134-40.
7.Nazer H, Roland J, Mowat A, et al. Wilson's disease in childhood. Clinical Pediatrics, vol.22, No.
11, Nov 1983.
8.Nazer H, Ede RJ, Mowat AP, et al. Wilson's's disease: clinical presentation and use of prognostic index.
Gut, 1986 27, 1377-1381.
9.Latif N, Mehmood K. Risk
factors for fulminant hepatic failure and their relation with outcome in
children. J Pak Med Assoc 2010 Mar; 60(3):175-8.
10.Tryambak S, Sumanta L, Radheshyam P, et al. Clinical profile, prognostic indicators and outcome of Wilson's’s
disease in children: a hospital based study. Trop Gastroenterol 2009; 30(3):
163-166.
11.Carter BA. Wilson's
disease. eMedicine Pediatrics: Genetics and Metabolic Disease. 2009; 9.
12.Lowette KF, Desmet K, Witters P, et al. Wilson's’s disease: long-term follow-up of a cohort of 24 patients
treated with D-penicillamine. Eur J Gastroenterol Hepatol 2010; 22(5):564-71.
13.Shah R, Piper MH. Wilson's disease. eMedicine
Gastroenterology Aug 25, 2009.
14.Kuloglu Z, Aydan K, Fuly D, et al. An unusual presentation of Wilson's’s disease in childhood nodular
fatty infiltration in liver. The Turkish Journal of Pediatrics 2008;
50:167-170.