ABSTRACT
The triad of hypoparathyroidism, sensori neural deafness, and variable renal system defects is known as HDR syndrome. It is an autosomal dominant inherited disorder. The clinical features of early onset deafness are the most completely penetrate part of this syndrome, but that of the hypoparathyroidism can present at any age. Here we report a 13-year-old Jordanian girl, presented with history of recurrent generalized tonic-clonic convulsions since two months, further history revealed bilateral hearing loss since birth. On examination, she has normal growth parameters, normal facial appearance. Her laboratory data were consistent with hypoparathyroidism. Radiological evaluations showed absent right kidney and basal ganglia’s calcifications. Diagnosis of HDR syndrome was made. In conclusion, congenital sensori neural deafness can be a guide for early diagnosis of HDR syndrome in order to start early management and prevention of central nervous system and renal system complications.
Key words: HDR syndrome, Hypoparathyroidism, Sensori neural deafness.
JRMS June 2015; 22(2): 59-62/ DOI: 10.12816/0011373IntroductionHDR Syndrome (Hypoparathyroidism-Sensori neural deafness and renal anomalies) is a genetic developmental disorder with clinical diversity.(1) The mode of inheritance is autosomal dominant,(2) and primarily caused by GATA3 haploinsufficency.(3) Ade novo heterozygous missense (c.815C→T) mutation, Thr272Ile, in exon 4 of the GATA3 gene was reported in a patient with HDR syndrome whose parents were negative for Thr272Ile.(4)The GATA3 gene is located at chromosome 10p14 and is non-overlapping with the DiGeorge clinical region at 10p13.(5,6) TheGATA3 protein is expressed in developing parathyroid glands, inner ears, and kidneys.(7,8) Abnormalities of this protein are more likely to result in two or more of the phenotypic features of the HDR Syndrome and not in one.(9) Here we report a 13year old female patient who is a known case of congenital sensori neural deafness presented with generalized tonic-clonic convulsion. Laboratory data showed hypocalcaemia, hyperphosphatemia, and low parathyroid hormone (PTH) level indicating hypoparathyroidism. Radiological evaluation showed absent right kidney, and basal ganglia’scalcifications. The triad of hypoparathyroidism, congenital sensori neural deafness and renal anomaly presented in our patient support the diagnosis of HDR Syndrome, which is the first case reported in Jordan.
Case ReportThirteen years-old-female patient presented to the emergency room with generalized tonic-clonic convulsion which was aborted spontaneously immediately upon arrival. She has history of two previous similar attacks within the previous two months without seeking medical advice by her parents. She was a product of premature labor (34 weeks gestational age), vaginal delivery, birth weight 1800gm, and was admitted in the NICU
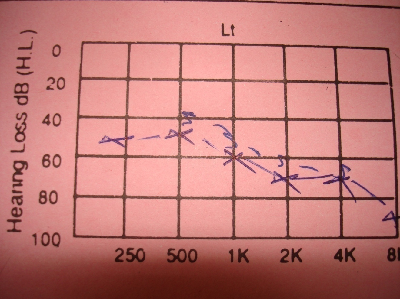
Fig. 1: Sensorineural deafness in left ear
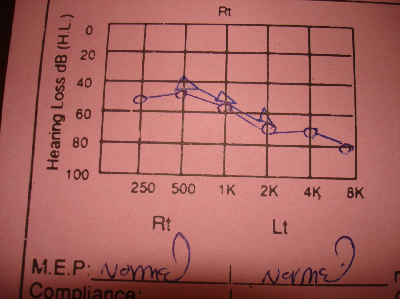
Fig. 2: Sensori neural deafness in right ear .
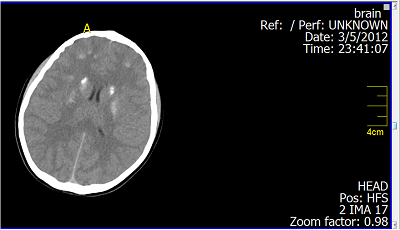
Fig. 3: Brain CT Scan: Basal ganglia calcifications.
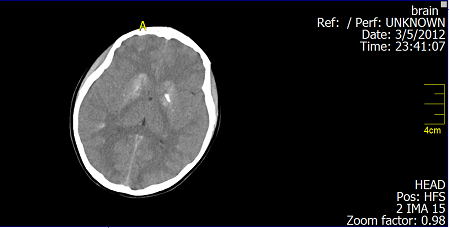
Fig. 4: Brain CT Scan: Basal ganglia calcifications
for two weeks with a picture of clinical sepsis. There was no history of use of ventilation or oxygen therapy. At age of one month, the mother noticed that her baby had decreased responses to high sounds and was diagnosed to have sensori neural deafness. Follow up developmental history was within normal limits. Parents are non consanguineous. She has three brothers. Her grandfather, father and one of her brothers (aged 4 year) were diagnosed to have isolated sensori neural deafness. Physical examination was normal, no dysmorphic features, no mucocutaneous fungal infection. Ophthalmological examination was normal. Hearing assessment showed mild to moderate sensori neural deafness. Laboratory data were, WBC 8600/ml, PCV 36%, MCV 77 fl, Platelets 270,000/ml, Ca++6.5mg/dl (N= 8.4-10.2), phosphorus level 8.6mg/dl (N= 2.8-5.4), PTH (N=15-65pg/ml), ALP 190 IU (N=<187 IU/L), HbA1c 5.6% (N= 4.2-6.2%), Free T4 1.2ng/dl (N=1.101.8ng/dL), Mg level 2.1mg/dl (N= 1.2-2.3 mg/dL), and Vit-B12 level 470pg/ml (N=200-550 pg/ml). Urinary Ca/Creatinine ratio 0.02 (N= <0.22mg/mg), Urine analysis and culture were free. Renal ultrasound and DMSA scan showed absent right kidney. Brain CT-scan showed basal ganglia’s calcifications. Skeletal survey was normal. Management with calcium capsules 500mg three times daily, and one-Alfa capsule 1mg twice daily was started.
DiscussionThe combination of hypoparathyroidism, hearing loss, and renal dysplasia was first described in 1977 by Barakat et al.(10) Our patient was diagnosed to have an isolated mild to moderate sensori neural deafness (Fig. 1, 2) in the neonatal period which was explained to be inherited from her father and grandfather through autosomal dominant inheritance. By the presented age (13 year), she started to have recurrent attacks of generalized tonic-clonic convulsions due to hypocalcaemia. Laboratory data showed also hyperphosphatemia and low parathyroid hormone indicating that the patient developed hypoparathyroidism. Her Brain CT-Scan showed basal ganglia’s calcifications (Fig. 3, 4), a finding of hypoparathyroidism, which may be a possible cause for the development of the convulsions. Adequate treatment of hypoparathyroidism may lead to marked clinical improvement of these findings.(11) The combination of congenital sensori neural deafness and hypoparathyroidism in our patient pointed to the diagnosis of HDR syndrome,
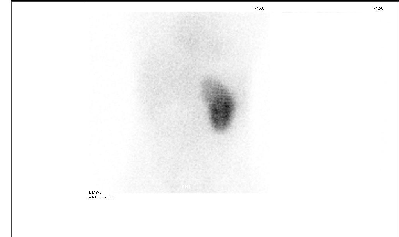
Fig. 5: DMSA Scan: Absent right kidney
especially as there were no features suggestive of hypoparathyroidism related to the polyglandular autoimmune syndrome such as mucocutaneous candidiasis or facial abnormalities that could be related to DiGeorge syndrome. Patients with HDR syndrome usually presents with hypocalcaemia, tetany, or afebrile convulsion at any age.(12) The clinical features of early onset deafness are the most completely penetrant part of the HDR syndrome, so that the diagnosis can be made by identifying the invariant hearing problem and the accompanying hypoparathyroidism not by genetic screen.(13) Our diagnosis was supported by the finding that the patient have absent right kidney on renal ultrasonic examination and DMSA Scan (Fig. 5), so that the clinical finding in our patient fulfilled the triad of HDR syndrome. Most studies showed that sensori neural deafness and hypoparathyroidism were present in all patients with HDR syndrome, but with variable expression of renal abnormalities and of parathyroid disorder, suggesting that additional genetic, environmental, or stochastic factors are likely involved in determining the clinical expression of this syndrome.(14) Renal disease includes nephrotic syndrome, cystic kidney, renal dysplasia, hypoplasia or aplasia, pelvicocalyceal deformity, vesicoureteral reflux, chronic renal failure, hematuria and renal scarring.(15) HDR syndrome without evidence of renal involvement was also reported.(16) A rare case of HDR syndrome was reported to have Hirschsprung disease but with no definite genetic association,(17) while another case with hypomagnesaemia due to the novel heterozygous deletion of two nucleotides (c.35-36delGC) in exon 2 of the GATA3 gene was reported in an Emirati patient.(18) Management of our patient with one-Alfa vitamin D and Calcium capsule showed cessation of the convulsions, but follow up brain CT-Scan 6 -12 month after treatment is needed.
Although our patient was diagnosed to have sensori neural deafness in the neonatal period, renal evaluation was not performed and a major anomaly was missed.
Conclusion Congenital sensori neural deafness can be a guide to evaluate the renal system and parathyroid function for the diagnosis of HDR syndrome, so that early management and prevention of central nervous complications or reflux nephropathy can be initiated.
AcknowledgmentSpecial thanks to Dr. Hiyam Shamoon for the help in diagnosing and reporting this case.
References1.
Coleman WB. Essential Concepts in Molecular Pathology, (1st ed). 2010; 462 .
2.
Bilous RW, Murty G, Parkinson DB, Thakker RV, et al. Autosomal dominant familial hypoparathyroidism, sensorineural deafness and renal dysplasia. N Engl J Med 1992; 327:1069-1074.
3.
Muroya K, Hasegawa T, Ito Y, Nagai T, et al. GATA3 Abnormalities and the phenotypic spectrum of HDR syndrome. J Med Genet 2001; 38: 374-380.
4.
Gomes T S, Gortner L, Dockter G, et al. HDR Syndrome - a follow-up genotype-phenotype analysis of a de novo Missense Thr 272Ile Mutation in Exon 4 of GATA3. Klin Padiatr 2012; 224 (7): 452-454 .
5.
Boyle DA. HDR Syndrome. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson Textbook of Pediatrics, 19th ed. Philadelphia, Elsevier Saunders, 2011.
6.
Lichtner P, Konig R, Hasegawa T, Van Esch H, et al. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J Med Genet 2000; 37: 33-37 .
7.
George KM, Leonard MW, Roth ME, Lieuw KH, et al. Embryonic expression and cloning of the murine GATA-3 gene. Development 1994; 120: 2673-2686.
8.
Viger RS, Guittot SM, Anttonen M, et al. Role of the GATA family of transcription factors in endocrine development, function, and disease. Molecular Endocrinology 2008; 22(4): 781-798.
9.
Ali A, Christie PT, Grigorieva IV, et al. Functional characterization of GATA3 mutation causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Human Molecular Genetics 2007; 16(3):
10.
Barakat AY, D'Albora JB, Martin MM, Jose PA. Familial nephrosis, nerve deafness, and hypoparathyroidism. J Pediatr 1977; 91(1): 61-64.
11.
Basak RC. A case report of basal ganglia calcification - a rare finding of hypoparathyroidism. OMJ 2009- 24, 220-222.
12.
Parks JS, Felner EI. Disorders of the hypothalamus and pituitary gland. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson Textbook of Pediatrics, 18th ed. Philadelphia, Elsevier Saunders 2007; p: 2291‐2293.
13.
Chiu w Y, Chen H W, Chao H W, et al. Identification of three novel mutations in the GATA3 Gene responsible for familial hypoparathyroidism and deafness in the chinese population. The Journal of Clinical Endocrinology and Metabolism 2006; 91(11): 4587-4592.
14.
Zahirieh A, Nesbit AM, Ali A, et al. Functional Analysis of a Novel GATA3 mutation in a family with the hypoparathyroidism, Deafness and Renal Dysplasia (HDR) Syndrome. Journal of Clinical Endocrinology & Metabolism first published February 10, 2005.
15.
Goodyer PR. Renal Dysplasia/ Hypoplasia. In: Avner E, Harmon W, Niaudet P. Pediatric Nephrology. Philadelphia, Lippincott Williams & Wilkins 2004; p: 83‐92 .
16.
Nanba K, Usui T, Nakamura M, et al. A Novel GATA3 Nonsense Mutation in a Newly Diagnosed Adult Patient of Hypoparathyroidism, Deafness, and Renal Dysplasia (HDR) Syndrome. Endocr Pract 2012 Nov 27:1-15 .
17.
Sephi MA, Behrouz B, Shooshtany FK. HDR syndrome (hypoparathyroidism, Sensorineural deafness and Renal disease). Iran J Pediatr 2010 March; 20(1):123-126.
18.
Al-Shibli A, Al Attrach I, WillemsP J. Novel DNA mutation in the GATA3 gene in an Emirati boy with HDR syndrome and hypomagnesemia. Pediatr Nephrol 2011; 26(7): 1167-1170 .