ABSTRACT
Thrombocytosis is a common feature of myeloproliferative neoplasms, including Philadelphia chromosome-positive chronic myeloid leukaemia. However, symptomatic extreme thrombocytosis causing thrombotic and/or haemorrhagic events is uncommon during the chronic phase of chronic myeloid leukaemia, especially when compared to Philadelphia chromosome-negative myeloproliferative neoplasms like polycythemia vera and essential thrombocythemia. We report a case of chronic myeloid leukaemia with a complicated clinical course. The patient did not show a molecular response to imatinib, a tyrosine kinase inhibitor, and was eventually admitted to the hematology/oncology department of King Hussein Medical Center with extreme thrombocytosis, prompting urgent plateletpheresis. Upon further investigation, molecular analysis to search for ABL kinase domain mutations revealed a positive result for the T315I mutation, explaining the resistance to treatment and disease progression.
Key words: Chronic myeloid leukemia; Extreme thrombocytosis; Tyrosine kinase inhibitor resistance; T315I mutation; Plateletpheresis
JRMS December 2019;26(3): 92-97 :10.12816/0054825
Introduction
Chronic myeloid leukaemia (CML) is a myeloproliferative neoplasm (MPN) which accounts for 15–20% of all cases of adult leukaemia.1 It is characterised by a genetic translocation t(9;22) which involves a fusion of the ABL1 gene on chromosome 9q34 with the BCR gene on chromosome 22q11.2. This forms the BCR-ABL fusion gene on the Philadelphia chromosome, which encodes a continuously activated non-receptor tyrosine kinase, resulting in uninhibited haematopoietic proliferation. 2-4
Patients diagnosed with this entity typically present with constitutional symptoms, anaemia and splenomegaly.Approximately 50% of patients are asymptomatic, and CML is discovered by an accidental reading of a high white cell count.5 Tyrosine kinase inhibitors (TKI) have revolutionised the therapeutic approach to CML. This is evident by an improvement in the 10-year survival rate from 20% to 80–90% with the first generation TKI, imatinib.6 Second and third generation TKIs have shown even higher potency in many studies.7-10 Response to treatment is best monitored by reverse transcription polymerase chain reaction (RT-PCR) of peripheral blood samples.11,12 Certain point mutations in the BCR-ABL kinase domain (KD) that confer resistance to one or more TKIs have been identified, and these mutations are associated with disease progression.13-15
We report a case of CML with extreme thrombocytosis, positive T315I mutation and resistance to multiple TKI treatments.
Case Report
We present the case of a 46-year-old female patient who was diagnosed with CML in the chronic phase in February 2015 with a platelet count of 800×103/µL. At the time of diagnosis, she had a low Sokal score of 0.7 and 95% positivity for the BCR-ABL fusion gene by fluorescence in situ hybridisation (FISH).
Frontline treatment with imatinib (400 mg) was initiated. In the beginning, a good response to treatment was evident as both haematological and cytogenetic responses were achieved. However, the patient had not achieved a molecular response as her RT-PCR result for the BCR-ABL fusion gene was never less than 2.7% over a 3-year follow-up period.
The dose of imatinib was increased to 600 mg, but the RT-PCR value increased to 25% in July 2018. At this point, a second-line treatment was considered, and the patient was switched to nilotinib at a dose of 400 mg twice daily. In her next visit to the clinic the following month, she complained of headache. A complete blood count revealed a very high platelet count of 4465 × 103/µL, which was confirmed by a peripheral blood smear (Figure 1).
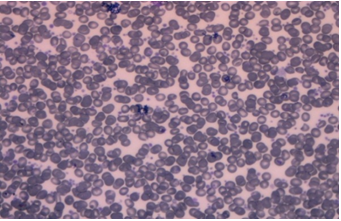
Figure 1. Peripheral blood smear showing extreme thrombocytosis.
The patient was immediately admitted to the hospital and started on hydroxyurea at a dose of 1 g twice daily, in addition to mechanical removal of platelets by plateletpheresis as a prophylactic measure. She underwent four sessions of platelet depletion using the Spectra Optia Apheresis System, lowering her platelet count to 1600 × 103/µL without any complication such as bleeding or hypocalcaemia. This was achieved over a period of 1 week, during which further investigations were carried out. Bone marrow study revealed granulocytic hyperplasia and marked megakaryocytic hyperplasia (Figure 2). FISH study showed positivity for the BCR-ABL fusion gene in 45% of cells. The diagnosis of CML in the accelerated phase was made according to the latest revision of the World Health Organization’s classification of myeloid neoplasms.16
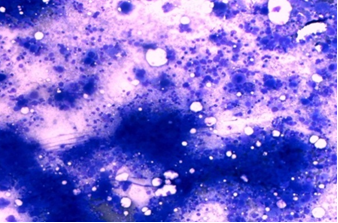
Figure 2. Bone marrow aspirate showing hypercellularity and a marked increase in megakaryopoiesis with micro-mononuclear (dwarf) forms.
The persistent finding of unresponsive extreme thrombocytosis merited further investigation, including JAK2 V617F, CALR , MPL and p-STAT5 mutations. Unfortunately, testing for only the first mutation was available, which revealed a negative result. A blood sample was sent to the hemostasis and thrombosis laboratory at Jordan University Hospital to look for BCR-ABL KD mutations. The result was positive for the T315I “gatekeeper” mutation. Ponatinib, a third generation TKI, is the drug of choice in such cases. However, at that time it was unavailable in Jordan, so the patient was started on a second generation TKI, dasatinib, at a dose of 100 mg per day. The dosage was reduced to 70 mg due to haematologic toxicity (anaemia and neutropenia), in addition to headache.
During regular follow-up at our hematology/oncology clinic in King Hussein Medical Center, the patient reported compliance to her medications, with no symptoms and no side-effects from the drug. The most recent hemogram performed in October 2019 revealed haematological response with a normal white cell count, as well as a normal platelet count of 308 × 103/µL. However, cytogenetic and molecular responses were not achieved, as her BCR-ABL levels assessed by FISH and RT-PCR were still 10% and 1.2%, respectively. The patient is planned to switch to ponatinib once it becomes available at our centre.
Discussion
The first generation TKI, imatinib, revolutionised the therapeutic approach to CML, evidenced by the decreased annual mortality rate of CML from 10–20% to 1–2%.17
Response to treatment is measured on three levels: hematologic, cytogenetic, and molecular. The molecular response has the greatest sensitivity for response monitoring, and is quantified by RT-PCR. A major molecular response is achieved when BCR-ABL falls to ≤0.1%.12 Our patient did not achieve a molecular response to imatinib, so was switched to nilotinib as a second-line treatment, after which her disease progressed to an accelerated phase with extreme thrombocytosis and a very high platelet count of 4465 × 103/µL. Disease progression despite second-line TKI treatment raised the possibility of additional genetic abnormalities. Therefore, BCR-ABL KD mutation analysis was performed, which revealed the presence of the T315I mutation.
In the era of TKIs, at least one in four patients will change TKI at least once due to intolerance or an inadequate response, i.e., drug resistance.11,14 Many point mutations in the BCR-ABL KD have been found to confer variable degrees of resistance.11-15 The most resistant among these is the T315I mutation, which is resistant to all of the currently approved TKIs except for ponatinib, the drug of choice for these patients.10,12 Unfortunately, we were unable to provide our patient with this drug at that time because it remained unavailable in Jordan.
Thrombocytosis is a poor prognostic factor of CML as it is associated with shorter survival.18 Thrombocytosis of more than 500 × 103/µL was reported by Jameel and Jamil in 10% of CML cases.19 However, extreme thrombocytosis exceeding 1000 × 103/µL is uncommon in CML, but is typical of Philadelphia chromosome-negative essential thrombocythemia (ET). Our patient had an extremely high platelet count higher than 4000 × 103/µL, which is rarely reported in the literature. Thus, an important differential diagnosis arose for our patient, which is reported in the literature as Philadelphia chromosome-positive ET.20,21 This entity was ruled out by the presence of features of CML in the peripheral blood and bone marrow in our case, which are typically absent in such ET cases. Also, molecular analysis for the JAK2 V617F mutation was performed, which revealed a negative result. Unfortunately, molecular studies for CALR, MPL and p-STAT5 mutations were not available.
Symptomatic extreme thrombocytosis is rarely described in CML case reports; however, the few available cases describe digital ischemia,22,23 myocardial infarction24 and neurologic symptoms such as uneasiness and headache, similar to our patient.22
According to Sora et al. and Liu Z, extreme thrombocytosis is more commonly seen in female CML patients, as seen here, with a median age of 59 years and high Sokal scores.25,26
Many studies explored different molecular mechanisms that result in the extreme thrombocytosis seen in CML patients. Lewandowski et al. found in their study that TKI-treated CML patients with thrombocytosis and positive JAK2 V617F or CALR mutation did not reach complete hematologic response due to the persistence of thrombocytosis. 27 Turakhia et al. found that p-STAT5 expression was activated in 85.7% of their CML patients with thrombocytosis. 28 Overexpression of EV11 gene was linked to extreme thrombocytosis in a reported case of CML.29
This rare case of CML highlights the importance of assessing the ABL KD mutation status and related molecular studies in cases that fail to respond to treatment. Determining the mutation profile of such patients is essential to provide the best and most cost-effective management, in addition to avoiding complications and disease progression, and prolonging event-free survival.
Abbreviations:
BCR-ABL KD BCR-ABL kinase domain
CML Chronic myeloid leukaemia
FISH Fluorescence in situ hybridisation
MPN Myeloproliferative neoplasm
RT-PCR Reverse transcription polymerase chain reaction
TKI Tyrosine kinase inhibitor
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2015; 65(1):5–29.
2. Rowley
JD. A new consistent chromosomal abnormality in chronic myelogenous
leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature
1973;243:290–3.
3. Shuai
K, Halpern J, ten Hoeve J. Constitutive activation of STAT5 by the
BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 1996;13:247–54.
4. Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer 2018;17:49.
5. Chang FF, Qazi RA, Khan M, Baloch S,
Sahito MM. Clinico
hematological profile and phase distribution of chronic myeloid leukemia. Biol
Med (Aligarh) 2015;7:5.
6. Deininger M, O’Brien SG, Guilhot F, et al. International randomized study of
interferon vs. STI571 (IRIS) 8-year follow up: sustained survival and low risk
for progression of events in patients with newly diagnosed chronic myeloid
leukemia in chronic phase (CML-CP) treated with imatinib. ASH 51st annual
meeting, 2009 Dec 5-8; New Orleans, Louisiana: Blood; 2009; p. 462.
7. Saglio G, Kim DW, Issaragrisil S, le
Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib
for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010;:2251–9.
8. Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala
M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase
chronic myeloid leukemia. N Engl J Med 2010; :2260–70.
9. Cortes JE, Gambacorti-Passerini C, Deininger MW, Mauro MJ,
Chuah C, Kim DW, et al. Bosutinib versus imatinib for newly diagnosed
chronic myeloid leukemia: results from the randomized BFORE trial. J Clin
Oncol 2018;:231–7.
10. Nicolini
FE, Basak GW, Kim DW, Olavarria E, Pinilla-Ibarz J, Apperley JF, et al. Overall
survival with ponatinib versus allogeneic stem cell transplantation in
Philadelphia chromosome-positive leukemias with the T315I mutation. Cancer 2017;123(15):2875–80.
11. Steegmann JL, Baccarani M, Breccia M,
et al. European
LeukemiaNet recommendations for the management and avoidance of adverse events
of treatment in chronic myeloid leukaemia. Leukemia 2016;30(8):1648–71.
12. Radich JP,
et al. Chronic myeloid leukemia, Version 1.2019, NCCN
clinical practice guidelines in oncology. J Natl Compr Canc
Netw 2018;16:1108–35.
13. Chaitanya PK, Kumar KA, Stalin B,
Sadashivudu G, Srinivas ML. The
role of mutation testing in patients with chronic myeloid leukemia in chronic
phase after imatinib failure and their outcomes after treatment modification:
single-institutional experience over 13 years. Indian J Med Paediatr Oncol
2017; 38(3):328–33.
14. Patel AB, O’Hare T, Deininger MW. Mechanisms of resistance to ABL kinase
inhibition in chronic myeloid leukemia and the development of next
generation ABL kinase inhibitors. Hematol Oncol Clin North Am
2017;31(4):589–612.
15. Wieczorek A, Uharek L. Management of chronic myeloid leukemia
patients resistant to tyrosine kinase inhibitors treatment. Biomark Insights
2016;10(Suppl 3):49–54.
16. Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumors of Hematopoietic and
Lymphoid Tissues. Revised 4th ed. Lyon: IARC; 2017.
17. Jabbour, E, Kantarjian, H. Chronic myeloid
leukemia: 2018 update on diagnosis, therapy and monitoring. Am J Hematol 2018;93:442–59.
18. Mason JE, Devita VT, Canellos GP. Thrombocytosis in chronic granulocytic
leukemia: incidence and clinical significance. Blood
1974;44:483–7.
19. Jameel A, Jamil SN. Clinico-pathological
profile of chronic myeloid leukemia. JPMI 2006; 20:235–8.
20. Hassankrishnamurthy
S, Mody MD, Kota VK. A
case of chronic myelogenous leukemia occurring in a patient treated for
essential thrombocythemia. Am J Case Rep 2019;20:10–4.
21. LeBrun DP, et al. Essential thrombocythemia with the
Philadelphia chromosome and BCR-ABL gene rearrangement. An entity distinct from
chronic myeloid leukemia and Philadelphia chromosome-negative essential
thrombocythemia. Cancer Genet Cytogenet 1991;54(1):21–5.
22. Thakral
B, Saluja K, Malhotra P, Sharma RR, Marwaha N, Varma S.
Plateletpheresis in a case of symptomatic thrombocytosis in chronic myeloid
leukemia. Ther Apher Dial 2004; 8:497–9.
23. Win N,
Mitchell DC. Platelet apheresis for digital gangrene due to
thrombocytosis in chronic myeloid leukaemia. Clin Lab Haematol 2001;23:65–6.
24. Ebrahem R, Ahmed B, Kadhem S, Truong Q. Chronic myeloid leukemia: a case of
extreme thrombocytosis causing syncope and myocardial infarction. Cureus
2016;8(2):e476.
25. Sora F, et al. Chronic myeloid leukaemia with extreme
thrombocytosis at presentation: incidence, clinical findings and outcome. Br J
Haematol 2017;181(2):267–70.
26. Zhihe Liu, Honqiong Fan, Yuying Li,
Chunshui Liu. Analysis of
clinical characteristics and efficacy of chronic myeloid leukemia onset with
extreme thrombocytosis in the era of tyrosine kinase inhibitors. Onco Target
Ther. 2017;10:3515-3520.
27. Lewandowski K, Gniot M, Wojtaszeska M,
Kandula Z, Becht R, Paczkowska E, et al. Coexistence of JAK2 or CALR mutation is a rare but
clinically important event in chronic myeloid leukemia patients treated with
tyrosine kinase inhibitor. Int Lab Hematol. 2018; 40(3):366-371.
28. Turakhia
SK, Murugesan G, Cotta CV, Theil KS. Thrombocytosis and STAT5 activation in
chronic
myelogenous leukaemia are not associated with JAK2 V617F or calreticulin
mutations. J Clin Pathol. 2016;69(8):713–719.
29. Balatzenko
G, Guenova M, Stoimenov A, Jotov G, Toshkov S. Philadelphia chromosome–positive chronic
myeloid leukemia with p190BCR-ABL rearrangement, overexpression of the EVI1 gene, and extreme
thrombocytosis: a case report. Cancer Genet Cytogenet. 2008;181(1):75–77.